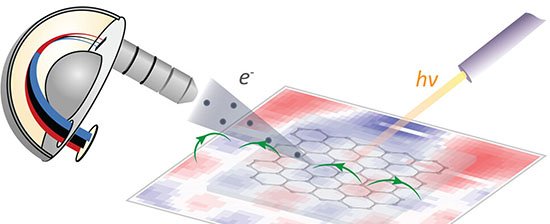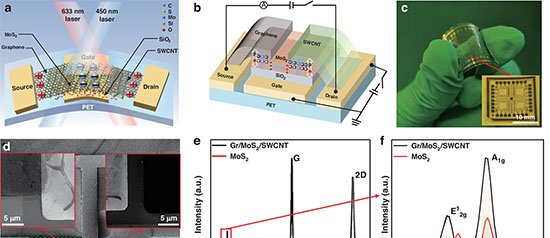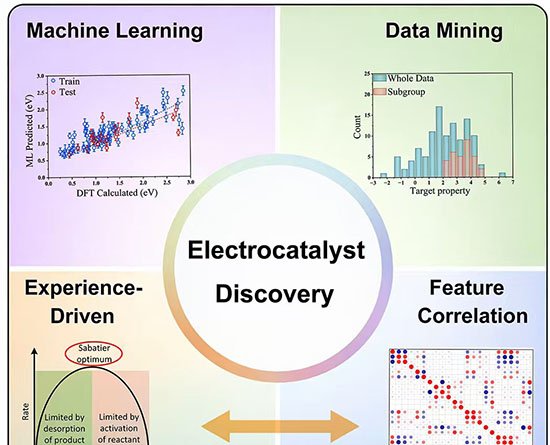
13 de fevereiro de 2026 – Pesquisadores da Universidade de Porto Rico e da Universidade Técnica de Munique apresentaram, em artigo publicado na revista Accounts of Materials Research, uma rota de descoberta de eletrocatalisadores que combina aprendizado de máquina interpretável e descritores fisicamente fundamentados. O método promete reduzir drasticamente o tempo de triagem de milhões de combinações químicas destinadas a reações essenciais para tecnologias de energia limpa, como divisão da água, redução de oxigênio em células a combustível, conversão de CO2 e fixação de nitrogênio.
Segundo o estudo, substituir metais nobres escassos – platina e irídio, entre outros – exige avaliar materiais formados por dezenas de elementos, diferentes estruturas cristalinas, dopantes variados e múltiplos suportes. Testar experimentalmente esse universo é inviável; já as abordagens computacionais tradicionais, baseadas em teoria do funcional da densidade (DFT) e em “gráficos vulcão”, simplificam demais sistemas que fogem ao comportamento de superfícies metálicas extensas, como catalisadores de átomo único.
Transparência como condição
Ao priorizar modelos que revelam suas próprias equações, a equipe defende que a interpretação direta dos resultados acelera a transição do computador para o laboratório. “A descoberta de catalisadores precisa de mais do que predição rápida; precisa transformar dados em compreensão”, afirmou o professor Zhongfang Chen, autor correspondente.
Três ferramentas complementares
O trabalho destaca um fluxo de análise global-para-local construído sobre três técnicas:
- LASSO (Least Absolute Shrinkage and Selection Operator) – algoritmo de regressão que elimina variáveis irrelevantes. Aplicado à reação de redução de nitrogênio em átomos metálicos únicos ancorados em lacunas de boro de diboretos bidimensionais, condensou parâmetros geométricos e eletrônicos em uma única fórmula capaz de prever a barreira de energia livre do passo limitante. O modelo indicou o sistema titânio em diboreto de vanádio como candidato promissor.
- SISSO (Sure Independence Screening and Sparsifying Operator) – forma novas combinações algébricas de descritores básicos. Para a redução de oxigênio em grafeno com átomos metálicos isolados, o método derivou um descritor bidimensional que une o centro da banda-d do metal e a energia de formação do suporte, mapeando tendências globais de atividade.
- Subgroup Discovery (SGD) – mineração de regras que identifica subconjuntos com desempenho fora da curva. Em estruturas metalorgânicas de níquel para evolução de oxigênio, o SGD definiu quatro condições relacionadas à ocupação orbital e à energia de ionização que sinalizam baixo sobretensão.
Do algoritmo ao laboratório
No estudo de mais de 10 000 catalisadores de átomo único em grafeno para redução de oxigênio, o SISSO delineou o panorama geral e o SGD isolou um subgrupo de alta performance. O melhor sistema – cobalto coordenado a dois átomos de enxofre e dois de nitrogênio – foi sintetizado e mostrou estabilidade prolongada, comprovando a eficácia da triagem.
Triagens em escala de banco de dados reforçaram a estratégia. De mais de 6 300 materiais listados na 2DMatPedia, filtros de sintetizabilidade, condutividade e energia de adsorção de oxigênio reduziram o conjunto a 26 catalisadores (24 para redução e dois para evolução de oxigênio) com atividades previstas comparáveis às de platina e óxido de irídio.
Outro exemplo usou aprendizado “progressivo” em duas etapas para identificar bons catalisadores em dados escassos. O modelo apontou manganês sobre dióxido de rutênio, depois validado experimentalmente com alta densidade de corrente e baixa sobretensão em meio ácido.
Para redução de CO2, a equipe avaliou 784 dímeros metálicos verticais (“sanduíche inverso”) sobre grafeno defeituoso. A diferença entre as primeiras energias de ionização dos dois metais emergiu como principal descritor, resultando em 154 candidatos estáveis e altamente ativos.

Imagem: Nanowerk https
Ligação entre simulação e experimento
Para confirmar previsões estruturais, um modelo supervisionado treinado em espectros simulados foi aplicado a dados de absorção de raios X de catalisadores de cobalto reais. A análise indicou alta concentração de sítios de borda coordenados a dois átomos de nitrogênio – as posições mais ativas observadas na prática.
Desafios e próximos passos
Modelar todo o interface eletroquímico – solvente, pH, dupla camada elétrica e potencial aplicado – continua sendo o principal obstáculo. Os autores sugerem cálculos de alta fidelidade em um subconjunto pequeno de materiais, seguidos de fusão multi-fidelidade com medições experimentais para gerar descritores consistentes em diferentes escalas.
Para o primeiro autor Liangliang Xu, a inteligência artificial caminha para atuar como parceira de pesquisa. Ele prevê agentes baseados em modelos de linguagem capazes de extrair protocolos de síntese, padronizar parâmetros e gerar listas de experimentos executáveis em laboratórios automatizados, encurtando o ciclo entre ideia e material validado.
Os casos reunidos no artigo mostram que, ao tornar visível o raciocínio dos algoritmos, a ciência de materiais aproxima a compreensão atômica de catalisadores reais para energias sustentáveis.
Com informações de Nanowerk